为什么FDA审核对国外医疗器械至关重要?
美国食品药品监督管理局(FDA)对医疗器械的监管被公认为全球最严格之一。通过FDA审核不仅意味着产品可以合法进入美国市场,更是打开加拿大、澳大利亚、日本等国家注册大门的“通行证”。**未通过FDA审核的产品,即使技术再先进,也只能被挡在北美市场之外**。
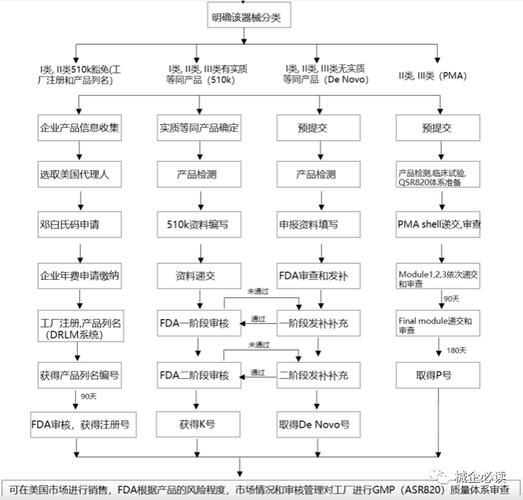
国外医疗器械注册流程拆解:从分类到510(k)的每一步
第一步:确认产品分类(Class I/II/III)
医疗器械在FDA体系中被分为三类,分类决定了后续路径:
- Class I(低风险):如压舌板、绷带,仅需企业注册与列名,豁免510(k)。
- Class II(中等风险):如血糖仪、电动轮椅,**必须提交510(k)文件**,证明与已上市器械实质等效。
- Class III(高风险):如心脏起搏器,需走最严苛的PMA(Premarket Approval)路径。
第二步:指定美国代理人(US Agent)
国外制造商必须指定一位**常驻美国的代理人**,负责与FDA沟通、接收官方文件。代理人需具备医疗器械法规背景,否则可能导致文件延误。
第三步:准备510(k)技术文档
510(k)的核心是“实质等效性”论证,需包含:
- 器械描述:材料、工作原理、预期用途。
- 对比器械:选择1-3个已获批的“谓词器械”,逐项对比性能数据。
- 生物相容性测试:ISO 10993系列报告,证明材料对人体无害。
- 软件验证:若含软件,需提交IEC 62304验证文档。
如何快速通过FDA审核?三大加速策略
策略一:提前进行Pre-Submission会议
在正式提交510(k)前,可申请与FDA的**Q-Sub会议**(费用约$300)。通过提前沟通,可明确:
- FDA对测试方案的认可意见;
- 是否需要额外临床数据;
- 标签说明书的修改建议。
**案例**:某德国血糖仪制造商因未提前沟通,被要求补充100例临床数据,导致审核周期延长6个月。

策略二:利用第三方审核机构(3P510k)
FDA授权了16家机构(如TÜV SÜD、BSI)进行**第三方510(k)审查**,优势包括:
- 审核时间缩短至30-60天(FDA直接审查需90-180天);
- 机构工程师更熟悉技术细节,反馈更清晰。
需注意:Class III器械、含药器械等**不适用于第三方路径**。
策略三:精准选择谓词器械
谓词器械的选择直接影响实质等效性论证的成功率。建议:
- 在FDA 510(k)数据库中筛选**近5年获批**的同类产品;
- 优先选择**适应症、技术特征高度匹配**的器械;
- 避免选择已被FDA发出安全警告的器械作为对比。
常见拒审原因与应对方案
拒审原因1:生物相容性测试不完整
问题:仅测试了细胞毒性,未进行皮肤刺激或致敏试验。
解决方案:按ISO 10993-1附录A的**器械接触时间矩阵**,补全所有必要测试。
拒审原因2:软件文档缺失
问题:未提供软件生命周期过程文档(如需求规范、缺陷管理记录)。
解决方案:遵循IEC 62304标准,补充**软件风险分析报告**和**可追溯性矩阵**。
拒审原因3:标签未标明禁忌症
问题:标签仅描述适应症,未列出“不可用于MRI环境”等禁忌症。
解决方案:参考FDA指南《Labeling for Medical Devices》,增加**黑框警告**。
国外制造商容易忽视的隐性成本
- 年度企业注册费:2024年FDA收费为$7,653,即使未销售也需缴纳。
- 美国代理人服务费:专业代理人年费约$5,000-$15,000,低价代理可能隐藏额外收费。
- 质量体系维护:通过510(k)后,FDA每2-3年进行一次**QSR(21 CFR 820)现场审核**,需持续投入合规成本。
2024年FDA审核的新变化
2024年10月起,FDA将强制要求510(k)提交使用**eSTAR模板**(电子化提交系统)。新模板包含:
- 结构化问答,减少自由文本描述;
- 自动校验功能,避免格式错误;
- 实时显示审核进度,降低沟通成本。
未使用eSTAR的提交将被直接拒绝接收。
评论列表