医疗器械注册到底要经历哪些阶段?
从立项到拿证,医疗器械注册通常被拆成六大阶段:立项评估→检验检测→临床评价→注册申报→技术审评→行政审批。每一步都有“坑”,提前规划才能把时间和预算压到最低。
(图片来源网络,侵删)
二类医疗器械备案与三类注册的核心差异
很多人把“备案”和“注册”混为一谈,其实二者在法规路径、资料深度、审批周期上完全不同。
- 法规依据不同:备案走《医疗器械监督管理条例》第三十条;注册走第三十一条。
- 审批机构不同:备案由设区市局当场办结;注册由国家局或省局审评中心技术审评。
- 周期差异:备案平均7个工作日;注册周期12-18个月。
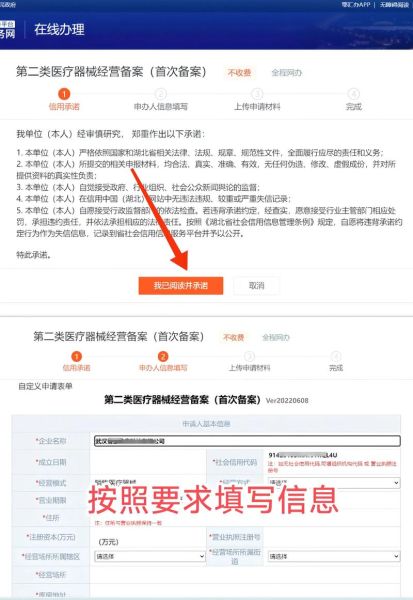
二类医疗器械备案条件逐条拆解
1. 主体资格:谁可以申请?
只能是境内生产企业或境外生产企业指定的境内代理人。贸易公司、经销企业无法以自己的名义备案。
2. 产品范围:哪些产品算二类?
以《医疗器械分类目录》为准,常见如电子血压计、制氧机、医用脱脂棉。不确定类别时,先做分类界定,避免后续退审。
3. 资料清单:缺一不可的七件套
- 产品风险分析资料
- 产品技术要求
- 产品检验报告(需在一年内)
- 临床评价资料(豁免临床的需提交豁免依据)
- 产品说明书及标签样稿
- 质量管理体系文件
- 营业执照及生产许可证复印件
注册检验最容易踩的四个坑
检验报告是备案/注册的核心材料,常见问题如下:
- 检测机构无资质:必须选择国家药监局认可的十大医疗器械检验中心。
- 样品批次不一致:送检样品必须与量产工艺一致,否则会被质疑真实性。
- 标准引用过期:检验依据必须是最新强制性国标或行标。
- EMC项目漏项:有源产品必须做电磁兼容,漏项直接退审。
临床评价路径怎么选?
二类器械多数可走豁免临床,但需满足《免于临床评价医疗器械目录》要求。如果不在目录内,有三条路:

(图片来源网络,侵删)
- 同品种比对:找已上市产品做对比,证明等同性。
- 临床试验:成本高,周期长,但数据最权威。
- 真实世界数据:通过上市后监测数据补充,适合已有销售基础的产品。
注册申报资料编写技巧
1. 产品技术要求怎么写?
按照《医疗器械产品技术要求编写指导原则》,必须包含性能指标、检验方法、术语定义。性能指标不能照抄标准,要结合产品设计输入。
2. 风险分析文件如何避坑?
采用ISO14971风险管理流程,输出风险管理报告。重点说明风险控制措施,例如血压计袖带漏气风险通过双气囊设计+报警提示降低。
审评发补常见问题TOP5
根据公开数据,发补率最高的五类问题:
- 说明书适应症描述超出备案范围
- 检验报告中关键项目缺项
- 临床评价资料无法证明安全有效
- 标签未注明禁忌症
- 生产地址与注册检验样品来源不一致
如何缩短整体周期?
三个实战技巧:
- 前置检验:在研发阶段同步送检,避免等产品定型后再排队。
- 预沟通:向省局审评中心申请预审查,提前发现资料硬伤。
- 模块化准备:把资料拆成技术模块+法规模块,同步推进。
境外企业备案特殊要求
境外生产企业需额外提交:

(图片来源网络,侵删)
- 境外上市证明(如CE、FDA)
- 中国境内代理人委托书(需公证认证)
- 境外质量体系认证(如ISO13485)
注意:所有外文资料必须中文翻译+公证。
备案成功后还要做什么?
备案不是终点,企业需在30日内建立上市后监测体系,包括:
- 不良事件收集流程
- 年度风险评价报告
- 产品追溯系统(UDI实施)
省局会按5%比例进行飞行检查,重点核查生产地址、设备、人员是否与备案资料一致。
评论列表