生物制品行业前景如何?一句话:全球需求持续攀升,中国市场年复合增长率有望保持两位数。生物制品注册流程是什么?先立项、再临床、后审评,拿到批件平均耗时三到五年。下面用问答式拆解,让你一次看懂。

(图片来源网络,侵删)
行业前景:为什么资本扎堆生物制品?
需求端:老龄化+慢病双重驱动
- **人口老龄化**:中国岁以上人口已超2.8亿,肿瘤、自身免疫病用药需求激增。
- **慢病年轻化**:糖尿病、银屑病等患者年龄下探,生物制剂疗效优于传统小分子。
供给端:技术迭代打开天花板
- **双抗、ADC、细胞基因治疗**三大前沿赛道融资热度不减,2023年全球ADC交易总额突破500亿美元。
- **国产替代**:PD-1单抗医保谈判后价格降幅超60%,本土企业市占率从不到10%跃升至45%。
政策端:审评提速+支付扩容
- 2020版《生物制品注册分类及申报资料要求》将临床试验默示许可从“排队”改为“默认制”,平均缩短8个月。
- 2023年国家医保目录新增111个药品,其中生物制品占比首次突破30%。
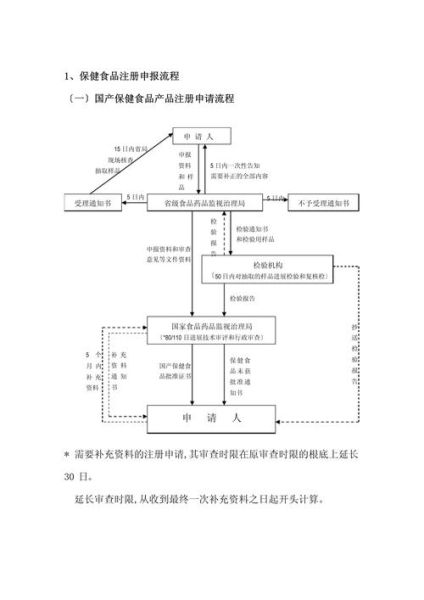
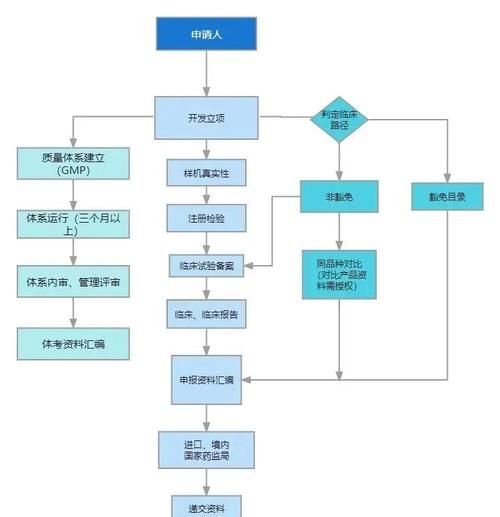
注册流程:从实验室到上市要闯几关?
第一步:立项与成药性评估
问:什么项目值得做?
答:先看**未满足的临床需求**,再看**技术壁垒**。以CAR-T为例,靶点CD19已红海,BCMA、Claudin18.2仍有机会。
第二步:临床前研究
- **药效学**:体外细胞杀伤、体内小鼠模型抑瘤率需达到统计学差异。
- **毒理学**:GLP实验室完成急性、重复给药、生殖毒性试验,周期12-18个月。
- **CMC**:细胞库三级管理、培养工艺放大至商业化规模,关键质量属性(CQAs)需锁定。
第三步:IND申报
问:资料包有多厚?
答:平均**8万页**,核心模块包括药学、药理毒理、临床方案。2023年NMPA受理IND申请同比增长37%,一次性通过率提升至78%。
第四步:临床试验
| 分期 | 目的 | 样本量 | 周期 |
|---|---|---|---|
| I期 | 安全性、剂量探索 | 20-100例 | 1-1.5年 |
| II期 | 初步疗效、适应症选择 | 100-300例 | 2-3年 |
| III期 | 确证性疗效、大样本安全 | 300-3000例 | 3-4年 |
第五步:NDA/BLA审评
- **优先审评**:罕见病、儿童用药可缩短至130个工作日。
- **现场核查**:生产现场动态检查,2023年通过率92%,主要缺陷集中在**无菌工艺验证**。
成本与风险:为什么90%项目倒在半路?
资金门槛
- 单抗:从IND到BLA平均投入**8-12亿元**。
- 细胞治疗:因个性化生产,成本上浮至**15-20亿元**。
技术风险
- **免疫原性**:ADA(抗药抗体)发生率超过30%即可能叫停。
- **病毒安全性**:逆转录病毒检测阳性曾导致某CAR-T项目III期失败。
政策变动
2024年《中国药典》拟新增**宿主细胞DNA残留量**限度从10ng/剂降至1ng/剂,多家在研企业需补做验证。
未来趋势:下一个风口在哪里?
多特异性抗体
PD-1/CTLA-4双抗已获批,**PD-1/LAG-3/TIGIT三抗**进入I期,理论可覆盖冷肿瘤。
体内基因编辑
相比体外CAR-T,**脂质纳米颗粒递送CRISPR**可降低成本70%,2023年首个体内编辑疗法获FDA批准。
(图片来源网络,侵删)
AI+CMC
利用机器学习预测细胞培养参数,**工艺开发周期从6个月压缩至6周**,药明生物已部署20+项目。
给从业者的三点建议
- **早布局专利**:靶点序列、制剂配方、工艺参数三维保护,避免上市后被狙击。
- **拥抱国际化**:中美双报可节省重复临床费用,2023年百济神州PD-1美国上市即采用中国临床数据。
- **关注支付创新**:按疗效付费、商业保险补充,解决高价生物药可及性难题。
(图片来源网络,侵删)
评论列表