体外诊断行业前景如何?
体外诊断(IVD)行业正处于高速扩张期。 **全球市场规模**:2023年已突破1100亿美元,预计2028年将达到1500亿美元,年复合增长率保持在6%左右。 **中国增速更快**:2023年国内市场规模约1300亿元人民币,2027年有望突破2000亿元,年复合增长率接近12%,显著高于全球平均。 自问自答:为什么中国跑得更快? 答: - **医保控费倒逼医院重视检验科创收**,检验项目外包比例持续上升; - **分级诊疗下沉**,基层医疗机构对快速、简便的POCT需求激增; - **带量采购只压耗材价不压服务费**,企业仍可通过高毛利试剂盈利。 ---体外诊断试剂注册流程
第一步:产品分类判定
- **三类风险最高**:如伴随诊断、基因检测试剂,需临床试验; - **二类中等风险**:如肿瘤标志物、激素检测试剂,可走同品种比对; - **一类低风险**:如采样拭子、清洗液,备案即可。 自问自答:如何快速判定类别? 答:登录国家药监局“医疗器械分类目录”检索关键词,或委托第三方法规事务所出具分类界定报告,**平均耗时5个工作日**。 ---第二步:注册检验与标准确认
- **送检机构**:必须为中国食品药品检定研究院(NIFDC)或省级检验所; - **检验周期**:二类试剂约3个月,三类试剂4–6个月; - **关键资料**:产品技术要求、说明书、性能评估报告、稳定性研究数据。 **加粗提醒**:若企业采用全新检测原理,需同步提交“非标方法学”验证资料,否则检验所会直接退审。 ---第三步:临床评价路径选择
- **临床试验**:三类试剂几乎必选,需三家三甲医院、不少于1000例样本; - **同品种比对**:二类试剂可对比已上市产品,重点证明“等效性”; - **豁免临床**:目录内部分项目如血型试剂,可直接提交文献资料。 自问自答:如何降低临床成本? 答: 1. **选择多中心联合PI**,共享剩余样本; 2. **采用真实世界数据(RWD)**,如医院LIS系统回顾性数据; 3. **提前与统计单位沟通**,避免方案设计缺陷导致样本量追加。 ---第四步:注册申报与审评审批
- **电子申报系统**:eRPS提交,格式为RPS ToC; - **技术审评**:二类60个工作日,三类90个工作日; - **发补概率**:首次通过率不足30%,常见发补项为“分析性能研究不充分”“说明书适应症过宽”。 **加粗技巧**:在发补回复中引用《体外诊断试剂注册申报资料要求》原文条款,可显著缩短二次审评时间。 ---第五步:生产许可与上市后监管
- **GMP核查**:注册批件后30日内申请,重点查“设计开发文档”与“批生产记录”一致性; - **唯一标识UDI**:2024年起所有三类试剂必须赋码; - **飞行检查**:每年抽查比例约5%,缺陷项集中在“留样管理”与“不良事件监测”。 自问自答:如何应对飞检? 答:建立“批号追溯链”,确保**每盒试剂可追溯到原料批号、生产操作人、检验放行记录**。 ---行业未来三大变量
变量一:集采扩围
- **安徽化学发光集采**:平均降价47%,但国产龙头装机量提升300%; - **下一步猜想**:分子诊断、伴随诊断或纳入省级联盟集采,**企业需提前布局“试剂+仪器”封闭系统**。 ---变量二:LDT模式合规化
- **北京、上海试点**:允许三甲医院开展“实验室自建项目”,无需注册证; - **影响**:高端特检项目(如甲基化早筛)可跳过注册直接盈利,**但数据需对接省级卫健委监管平台**。 ---变量三:出海合规门槛
- **欧盟IVDR**:2024年5月26日全面强制,技术文档要求提升10倍; - **美国FDA 510(k)**:需证明“实质等效”,但伴随诊断必须走PMA,耗时18个月; - **新兴市场**:东南亚接受CE认证,**注册成本仅为国内1/3**。 ---企业实战建议
- **产品线规划**:优先布局“集采免疫+高端分子”双轮驱动,避免单一赛道价格战; - **注册团队配置**:法规、医学、统计、生产四部门协同,**三类项目至少提前两年启动**; - **现金流管理**:临床试验费用占注册成本60%,可通过“里程碑付款”外包给CRO降低风险。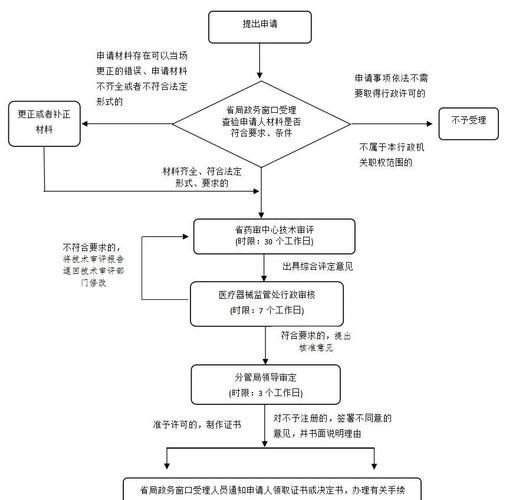
(图片来源网络,侵删)
评论列表