临床试验流程到底怎么走?
很多人以为“临床试验”只是医生给患者试新药,其实它是一套被全球监管机构严格定义的**标准化流程**。下面用通俗语言拆解,让你一次看懂。
(图片来源网络,侵删)
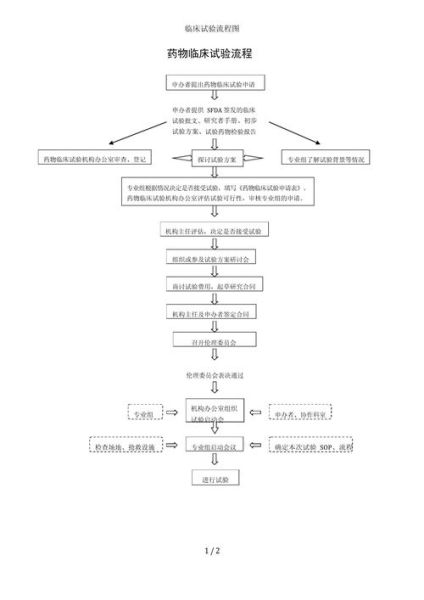
1. 试验启动前:伦理与法规双重把关
- 立项申请:药企或医院向国家药监局(NMPA)提交临床试验申请(IND)。
- 伦理审查:医院伦理委员会评估风险收益比,**保护受试者权益**。
- 注册公示:在中国需在“药物临床试验登记与信息公示平台”公开信息。
2. 试验分期:I期到IV期到底差在哪?
| 分期 | 受试者人数 | 核心目的 |
|---|---|---|
| I期 | 20–100人 | 观察**人体耐受性**与初步药代动力学 |
| II期 | 100–300人 | 初步验证**疗效**并继续评估安全性 |
| III期 | 300–3000人 | 大规模确认疗效,**对照现有标准治疗** |
| IV期 | 上市后数千至数万 | 长期**真实世界**安全性监测 |
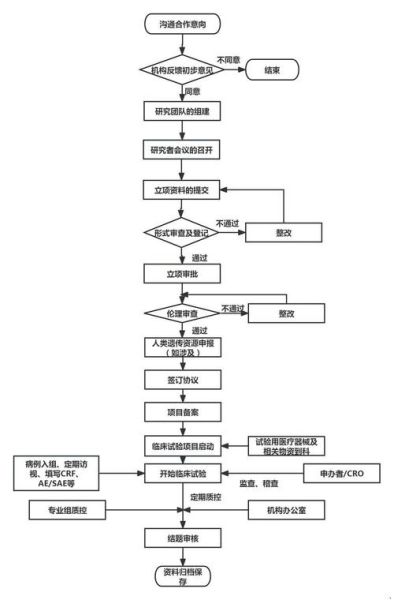
3. 受试者招募:谁能参加?
常见入选标准
- 年龄、性别、疾病分期符合方案要求
- 既往治疗失败或标准治疗无效
- 重要器官功能指标在允许范围内
常见排除标准
- 怀孕或哺乳期女性
- 合并严重心肝肾功能不全
- 近三个月参加过其他临床试验
临床试验费用是多少?
答案:国内III期肿瘤药单例成本约**30–50万元**,整个项目总费用可达**1–5亿元**,具体因适应症、中心数量、随访时长而异。
1. 费用构成拆解
- 受试者补贴:交通、营养、误工费,单例5000–20000元不等
- 检查费用:实验室、影像、基因检测,单例约5–10万元
- 研究者费用:医院及医生劳务费,占预算15–25%
- 药品生产与物流:GMP车间制备、冷链运输,占预算10–20%
- 数据管理与统计:EDC系统、第三方统计,占预算5–10%
2. 为什么不同适应症差距巨大?
以**罕见病**为例,患者分散、招募困难,单例成本可能飙升至**80–100万元**;而**高血压**这类常见病,患者基数大,单例成本可压到**10万元以内**。
3. 如何降低试验费用?
药企与CRO(合同研究组织)常用三大策略:
(图片来源网络,侵删)
- 去中心化试验(DCT):通过可穿戴设备远程采数据,减少现场随访
- 适应性设计:期中分析后调整样本量,避免过度招募
- 真实世界数据补充:利用医保数据库替代部分对照组随访
受试者最担心的五个问题
Q1:参加试验是不是当“小白鼠”?
不是。所有试验药物均已完成**体外实验、动物毒理实验**,且伦理委员会全程监督,受试者可随时退出。
Q2:试验药物一定免费吗?
国内规定**试验用药及方案要求的检查**必须免费;若出现试验相关损害,申办方需承担治疗费用。
Q3:会不会分到安慰剂组耽误病情?
肿瘤领域多为**“对照组接受标准治疗”**,而非安慰剂;若标准治疗失败,可申请交叉用药。
Q4:个人信息会泄露吗?
数据采用**匿名编码**,姓名、身份证号不会出现在任何公开报告中。
Q5:试验结束后还能继续用药吗?
若药物有效,申办方可提供**扩展用药计划(EAP)**,直至上市或患者不耐受。
(图片来源网络,侵删)
企业如何控制预算超支?
根据2023年《中国临床试验成本白皮书》,超支原因前三位:
- 患者招募延迟(占42%)
- 方案违背导致数据剔除(占28%)
- 中心启动滞后(占15%)
解决方案:
- 预筛数据库:提前锁定潜在患者,缩短招募周期
- 线上培训:减少研究者对方案的误解
- 风险监查(RBM):聚焦高风险中心,降低现场监查成本
未来趋势:数字化如何重塑成本结构?
AI辅助影像判读可节省**30%影像读片费用**;区块链技术确保数据不可篡改,减少**10–15%稽查成本**;电子知情同意(eConsent)让偏远地区患者也能参与,**扩大招募半径**。
随着国家药监局加入ICH,中国数据被全球认可,跨国药企更愿意在中国同步开展早期试验,**国内CRO报价有望与国际接轨**,进一步压缩利润空间,但对患者而言,意味着**更早用上全球最新疗法**。
评论列表