一、植入医疗器械行业前景如何?
市场规模持续扩大:全球植入医疗器械市场已从2020年的约1200亿美元增长至2023年的1450亿美元,年复合增长率保持在6%左右。中国作为第二大单体市场,2023年规模突破900亿元人民币,增速高于全球平均水平。
老龄化与慢病双重驱动:60岁以上人口比例预计2035年将达28%,关节、心脏、神经刺激类植入物需求同步上升。
技术升级带来新蓝海:可降解镁合金支架、闭环神经调控器、3D打印个性化植入体等创新产品正加速临床转化。
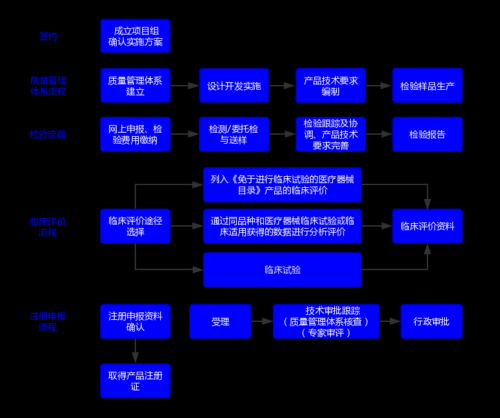
二、植入医疗器械注册流程
1. 产品分类先搞清:三类还是二类?
自问:我的植入物到底算哪一类?
自答:凡是“长期植入人体”且“潜在风险高”的,几乎都被归为第三类医疗器械,需走国家药监局(NMPA)审批通道;部分短期接触骨组织的可吸收螺钉可能被归为第二类,由省级局审批。分类目录每年动态调整,务必以最新文件为准。
2. 临床前研究要做哪些实验?
- 生物相容性:细胞毒性、致敏、刺激、遗传毒性、植入试验(ISO 10993系列)
- 力学性能:疲劳、磨损、腐蚀、微动腐蚀(ASTM F138、ISO 5832)
- 动物实验:大动物骨植入模型验证骨整合与降解速率
- 灭菌验证:EO残留、辐射灭菌剂量确认(ISO 11137)
3. 临床试验设计关键点
是否必须做临床试验?
若产品列入《免于临床试验目录》且能证明等同性,可提交临床评价报告;否则需开展前瞻性、多中心、随机对照试验。
样本量怎么算?
以骨钉为例,非劣效界值设为5%,α=0.05,β=0.2,预期成功率95%,每组至少需92例,考虑脱落率20%,总样本量220例左右。
临床终点怎么选?
骨科植入物常用术后6个月骨愈合率;心脏起搏器则关注12个月无并发症生存率。
4. 注册申报资料清单
- 申请表(eRPS系统在线填报)
- 产品技术要求(含性能指标与检验方法)
- 风险分析报告(ISO 14971)
- 临床评价资料或临床试验报告
- 生产制造信息(工艺流程图、关键工艺验证)
- 产品说明书与标签样稿
- 质量管理体系核查报告(GMP现场检查)
5. 审评审批时间线
| 阶段 | 法定时限 | 实际平均耗时 |
|---|---|---|
| 受理 | 5个工作日 | 3个工作日 |
| 技术审评 | 90个工作日 | 120-150个工作日 |
| 补正资料 | 1年内完成 | 平均补正2次,耗时6个月 |
| 行政审批 | 20个工作日 | 15个工作日 |
总周期:18-24个月;创新通道可缩短至12个月。
三、企业如何提前布局?
1. 建立注册前置团队
法规事务(RA):实时跟踪NMPA指南更新,提前识别分类变化风险。
质量保证(QA):在研发阶段即植入GMP理念,减少后期整改。
临床事务(CRA):与PI建立长期合作,缩短伦理审批时间。
2. 资金与时间双保险
- 注册预算占研发总投入的25%-30%,含检测、临床、审评费
- 设置20%不可预见费应对补正、发补、体系核查整改
- 采用分段式里程碑付款外包CRO,降低一次性资金压力
3. 国际化同步策略
CE-MDR与FDA PMA并行:欧盟2027年全面强制执行MDR,技术文件要求趋严;FDA对高风险植入物要求真实世界数据(RWD)。
利用QSR 820与ISO 13485差异:设计变更控制、CAPA体系提前对齐,避免重复审核。
东南亚跳板:新加坡HSA、马来西亚MDA审批周期短,可作为海外市场首发地,积累临床数据反哺中国注册。
四、常见误区与避坑指南
误区一:等同性论证随便写
错误做法:仅对比材料成分,忽略力学性能、表面改性、灭菌方式差异。
正确姿势:按照《医疗器械临床评价技术指导原则》逐一对比适用范围、技术特征、生物学特性,差异部分需补充台架或动物试验。
误区二:临床试验机构随便选
错误做法:只看伦理审批速度,忽视机构资质与PI经验。
正确姿势:优先选择国家药监局备案的临床试验机构,PI需具备同类植入物研究经历,且机构具备影像评估中心实验室。
误区三:忽视上市后监管
风险:注册证获批后若发生严重不良事件未及时上报,可能被暂停销售。
对策:建立上市后监测(PMS)系统,每季度汇总不良事件趋势,主动开展用户培训与再评价。
评论列表